 English
English  Ελληνικά
Ελληνικά 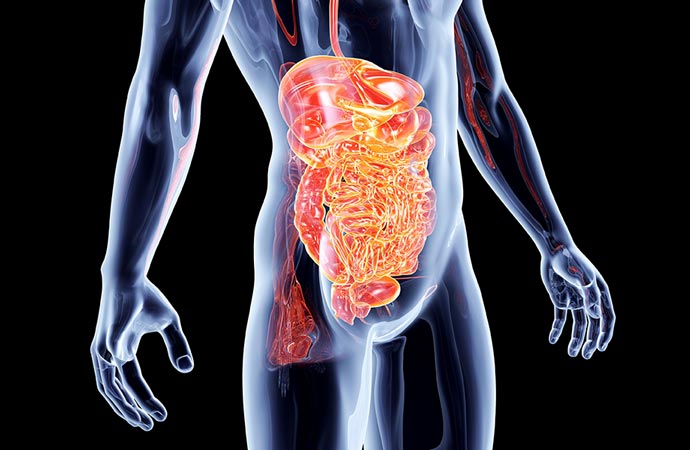
GIST gastrointestinal stromal tumors represent 0.2% of all neoplasms and 80% of sarcomas of the gastrointestinal system. It was originally thought to be derived from Cajal's intermediate cells, which are located in the Auerbach nerve plexus and are called the pacemaker of the gastrointestinal system. But new data show that they come from pluripotent stem cells.
Patients with GIST are usually over the age of 50 but they can emerge in younger people and are slightly more common in men. They develop along the entire length of the gastrointestinal tract, 60% are located in the stomach, 30% in the small intestine, 5% in the large intestine and the remaining 5% in other locations such as the rectum, esophagus, mesentery and retroperitoneum.
Most GISTs are sporadic, non-hereditary and have no clear cause. In rare cases they have been found in many members of the same family because these members inherited a gene mutation that could lead to GIST. Such syndromes are Familiar Gastrointestinal Stromal Tumor Syndrome, Neurofibromatosis type 1 and Carney-Stratakis Syndrome.
They are usually asymptomatic tumors that are found accidentally on endoscopic or laparoscopic examination. Depending on their location and nature (cellularity, profuse perfusion) they may present with symptoms of bleeding (hematemesis, black stools, anemia) or with symptoms of obstruction.
They usually emerge as solitary and rarely as multiple lesions and can develop either endoluminally or extraluminally to adjacent structures. They metastasize mainly hematogenously mainly in the liver and rarely in the lymph nodes. A special form of metastasis is the intra-abdominal dispersal which carries a high risk of recurrence or metastasis throughout the venter.
The diagnosis is based on computerized axial tomography (CAT) and magnetic resonance imaging (MRI). PET (Positron Emission Tomography) is sometimes necessary to stage and monitor treatment. Endoscopy (gastroscopy, colonoscopy) is performed to diagnose tumors in the upper and lower digestive tract and can be supplemented with endoscopic ultrasound and possibly a biopsy.
Histologically we distinguish two types, the atractoids that represent 70% and the epithelioids that represent 20%. The remaining 10% corresponds to the mixed type where we see both atractoid and epithelial elements.
The majority of GISTs have activated mutations in the c-kit gene. The mutation produces a substance (ligant) that causes uncontrolled activation of tyrosine kinase which in turn causes uncontrolled cell growth. In about 5% there is a mutation in the PDGFR-a gene. Mutation in these genes predicts response to tyrosine kinase inhibitor therapy (imatinib, Glivec).
Surgical Ro resection with sometimes en block resection of adjacent organs is the treatment of choice and should be sought in the first operation by avoiding rupture of the pseudocapsule of the tumor leading to dispersal and implantation. Postoperatively the patient is treated with imatib (Glivec). Major primary GISTs should undergo neoadjuvant chemotherapy with imatib and surgery in the second year. Also for the treatment of metastases treatment of choice is chemotherapy with a tyrosine kinase inhibitor. Laparoscopic GIST resection is generally indicated and has no difference in patient survival in the various studies.
Recently in the treatment of GISTs in addition to imatinib (Glivec) sunitib (Sutend) has been added and in clinical trials are three other drugs, nilitinib (Tasigna), everolimus and AMG 707.